Data splitters
Splitting your data in a Machine Learning pipeline is a crucial step. It is important to make sure that the data is split in a way that the model is not overfitting. In this tutorial we will show you how to use the splitters in DeepMol. Some splitters allow us to have an easier task for a model to learn, while others allow us to have a more difficult task for a model to learn. We will show you how to use the splitters and how to use them in a pipeline.
Create function to generate t-SNE embeddings
from rdkit import Chem, DataStructs
from rdkit.Chem import AllChem
import numpy as np
from sklearn.manifold import TSNE
import matplotlib.pyplot as plt
def generate_tsne_molecular_similarities(train_dataset, valid_dataset, test_dataset):
# Create a function to compute molecular fingerprints
def compute_fingerprint(smiles):
molecule = Chem.MolFromSmiles(smiles)
fingerprint = AllChem.GetMorganFingerprintAsBitVect(molecule, 2, nBits=1024)
return fingerprint
# Compute molecular fingerprints for each molecule in the combined dataset
fingerprints = [compute_fingerprint(smiles) for smiles in train_dataset.smiles]
fingerprints.extend([compute_fingerprint(smiles) for smiles in valid_dataset.smiles])
fingerprints.extend([compute_fingerprint(smiles) for smiles in test_dataset.smiles])
similarity_matrix = np.zeros((len(fingerprints), len(fingerprints)))
# Compute molecular similarities using Tanimoto coefficient
for i in range(len(fingerprints)):
for j in range(i + 1, len(fingerprints)):
similarity = DataStructs.TanimotoSimilarity(fingerprints[i], fingerprints[j])
similarity_matrix[i, j] = similarity
similarity_matrix[j, i] = similarity
# Apply t-SNE to reduce the dimensionality
tsne = TSNE(n_components=2, random_state=42)
tsne_embeddings = tsne.fit_transform(similarity_matrix)
# Separate the embeddings based on the original datasets
dataset1_embeddings = tsne_embeddings[:len(train_dataset.smiles)]
dataset2_embeddings = tsne_embeddings[len(train_dataset.smiles):len(valid_dataset.smiles) + len(train_dataset.smiles)]
dataset3_embeddings = tsne_embeddings[len(valid_dataset.smiles) + len(train_dataset.smiles):]
# increase the size of the plot
plt.figure(figsize=(10, 10))
# Plot the embeddings with different colors for each dataset
plt.scatter(dataset1_embeddings[:, 0], dataset1_embeddings[:, 1], c='red', label='Train dataset', s=[2]*len(dataset1_embeddings))
plt.scatter(dataset2_embeddings[:, 0], dataset2_embeddings[:, 1], c='blue', label='Validation dataset', s=[2]*len(dataset2_embeddings))
plt.scatter(dataset3_embeddings[:, 0], dataset3_embeddings[:, 1], c='green', label='Test dataset', s=[2]*len(dataset3_embeddings))
plt.legend()
plt.xlabel('t-SNE Dimension 1')
plt.ylabel('t-SNE Dimension 2')
plt.title('Molecular Similarities with t-SNE')
plt.show()
from deepmol.loaders import CSVLoader
dataset = CSVLoader("../data/CHEMBL217_reduced.csv", id_field="Original_Entry_ID",
smiles_field="SMILES", labels_fields=["Activity_Flag"]).create_dataset()
SingletaskStratifiedSplitter
A single task stratified splitter splits the data into train, validation and test sets. The data is split in a way that the distribution of the labels is the same in each set. This is useful when we have a dataset with a large number of classes or a highly unbalanced dataset, and we want to make sure that the distribution of the classes is the same in each set.
from deepmol.splitters import SingletaskStratifiedSplitter
splitter = SingletaskStratifiedSplitter()
train_dataset, valid_dataset, test_dataset = splitter.train_valid_test_split(dataset, frac_train=0.8, frac_valid=0.1, frac_test=0.1)
Below we can see the distribution of the labels in each set
As you might see, the distribution of the labels is roughly the same in each set.
train_dataset.get_shape()
((13316,), None, (13316,))
import pandas as pd
pd.Series(train_dataset.y).value_counts()
0 6658
1 6657
dtype: int64
valid_dataset.get_shape()
((1664,), None, (1664,))
import pandas as pd
pd.Series(valid_dataset.y).value_counts()
1 849
0 832
dtype: int64
test_dataset.get_shape()
((1665,), None, (1665,))
import pandas as pd
pd.Series(test_dataset.y).value_counts()
0 833
1 816
dtype: int64
Below we can see the distribution of the molecules in each set
generate_tsne_molecular_similarities(train_dataset, valid_dataset, test_dataset)
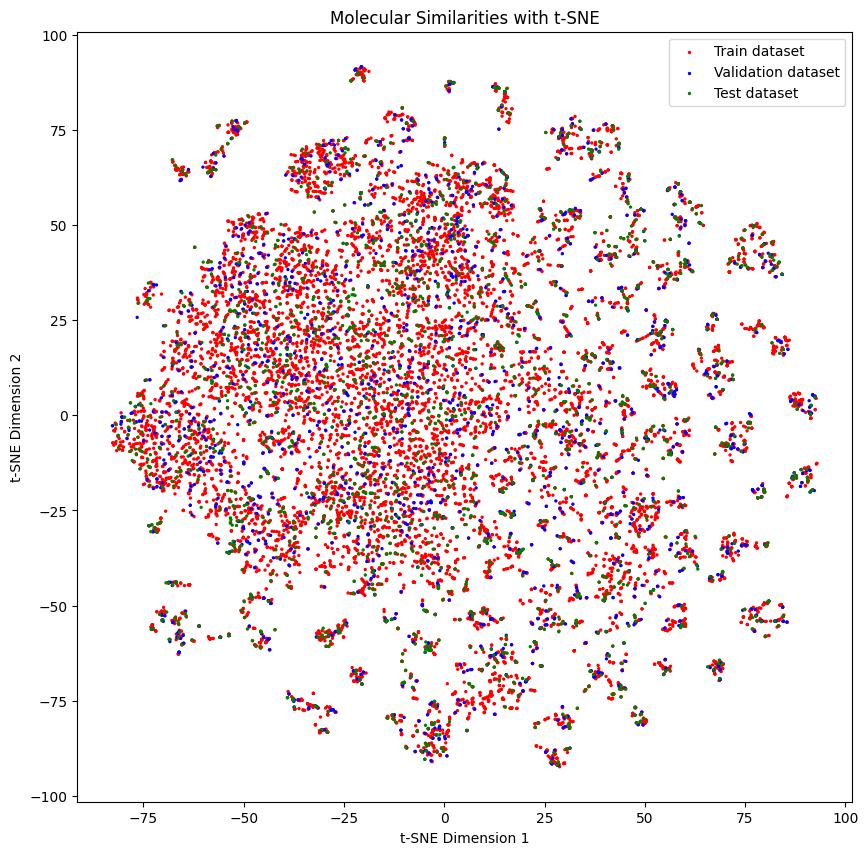
Then you can train them with a model. Here we use a random forest classifier.
from deepmol.compound_featurization import MorganFingerprint
from deepmol.models import SklearnModel
from deepmol.metrics import Metric
from sklearn.ensemble import RandomForestClassifier
from sklearn.metrics import accuracy_score
MorganFingerprint().featurize(train_dataset, inplace=True)
MorganFingerprint().featurize(valid_dataset, inplace=True)
MorganFingerprint().featurize(test_dataset, inplace=True)
rf = RandomForestClassifier()
model = SklearnModel(model=rf)
model.fit(train_dataset)
model.evaluate(test_dataset, [Metric(accuracy_score, name="accuracy")])
({'accuracy': 0.9843843843843844}, {})
from deepmol.metrics import Metric
model.evaluate(valid_dataset, [Metric(accuracy_score, name="accuracy")])
({'accuracy': 0.9898869720404521}, {})
RandomSplitter
from deepmol.splitters import RandomSplitter
train_dataset, valid_dataset, test_dataset = RandomSplitter().train_valid_test_split(dataset, frac_train=0.8, frac_valid=0.1, frac_test=0.1)
MultiTaskSplitter
from deepmol.loaders import CSVLoader
dataset = CSVLoader("../data/tox21.csv", id_field="mol_id",
smiles_field="smiles", labels_fields=["NR-AR","NR-AR-LBD","NR-AhR",
"NR-Aromatase", "NR-ER", "NR-ER-LBD",
"NR-PPAR-gamma", "SR-ARE","SR-ATAD5","SR-HSE","SR-MMP","SR-p53"]).create_dataset()
[18:35:38] WARNING: not removing hydrogen atom without neighbors
2023-06-27 18:35:39,339 — INFO — Assuming multitask since y has more than one dimension. If otherwise, explicitly set the mode to 'classification' or 'regression'!
from deepmol.splitters import MultiTaskStratifiedSplitter
train_dataset, test_dataset = MultiTaskStratifiedSplitter().train_test_split(dataset, frac_train=0.8)
import numpy as np
num_ones_per_column_train_dataset = np.sum(train_dataset.y == 1, axis=0)
num_ones_per_column_test_dataset = np.sum(test_dataset.y == 1, axis=0)
total = num_ones_per_column_train_dataset + num_ones_per_column_test_dataset
print("The percentage of each task label in the training set is:", num_ones_per_column_train_dataset / total)
print("The average percentage of the tasks labels in the training set is :", np.mean(num_ones_per_column_train_dataset / total))
print()
print("The percentage of each task label in the test set is:", num_ones_per_column_test_dataset / total)
print("The average percentage of the tasks labels in the test set is :", np.mean(num_ones_per_column_test_dataset / total))
The percentage of each task label in the training set is: [0.77669903 0.79746835 0.79427083 0.78333333 0.80075662 0.80857143
0.8172043 0.79087049 0.74242424 0.7983871 0.82244009 0.79196217]
The average percentage of the tasks labels in the training set is : 0.7936989991588942
The percentage of each task label in the test set is: [0.22330097 0.20253165 0.20572917 0.21666667 0.19924338 0.19142857
0.1827957 0.20912951 0.25757576 0.2016129 0.17755991 0.20803783]
The average percentage of the tasks labels in the test set is : 0.20630100084110578
from deepmol.splitters import RandomSplitter
train_dataset, test_dataset = RandomSplitter().train_test_split(dataset, frac_train=0.8)
import numpy as np
num_ones_per_column_train_dataset = np.sum(train_dataset.y == 1, axis=0)
num_ones_per_column_test_dataset = np.sum(test_dataset.y == 1, axis=0)
total = num_ones_per_column_train_dataset + num_ones_per_column_test_dataset
print("The percentage of each task label in the training set is:", num_ones_per_column_train_dataset / total)
print("The average percentage of the tasks labels in the training set is :", np.mean(num_ones_per_column_train_dataset / total))
print()
print("The percentage of each task label in the test set is:", num_ones_per_column_test_dataset / total)
print("The average percentage of the tasks labels in the test set is :", np.mean(num_ones_per_column_test_dataset / total))
The percentage of each task label in the training set is: [0.82200647 0.82700422 0.80729167 0.82333333 0.80075662 0.85142857
0.82795699 0.80785563 0.83712121 0.7983871 0.81045752 0.8108747 ]
The average percentage of the tasks labels in the training set is : 0.818706169088316
The percentage of each task label in the test set is: [0.17799353 0.17299578 0.19270833 0.17666667 0.19924338 0.14857143
0.17204301 0.19214437 0.16287879 0.2016129 0.18954248 0.1891253 ]
The average percentage of the tasks labels in the test set is : 0.18129383091168402
As you see the MultiTaskSplitter ensures the stratification of the classes in the training and test sets, while the RandomSplitter does not. Although the difference is not big, the multi-task splitter is more robust and scalable for extremely labelled datasets
Similarity Splitter
A similarity splitter splits the data into train, validation and test sets. The data is split in a way that the similarity between the molecules in each set is below a certain threshold. This is useful when we want to make sure that the molecules in the validation and test sets are either not too similar or similar to the molecules in the training set.
This splitter considers all the compounds with a similarity lower than the homogenous_threshold to be in the same set. The higher the threshold is, the more heterogeneous the split will be.
Moreover, this method tries to ensure the stratification of classes.
from deepmol.splitters import SimilaritySplitter
similarity_splitter = SimilaritySplitter()
train_dataset, valid_dataset, test_dataset = similarity_splitter.train_valid_test_split(dataset, frac_train=0.8, frac_valid=0.1, frac_test=0.1, homogenous_threshold=0.7)
Below we can see the distribution of the labels in each set
import pandas as pd
pd.Series(train_dataset.y).value_counts()
0 6678
1 6638
dtype: int64
import pandas as pd
pd.Series(valid_dataset.y).value_counts()
1 843
0 821
dtype: int64
import pandas as pd
pd.Series(test_dataset.y).value_counts()
1 841
0 824
dtype: int64
Below we can see the distribution of the molecules in each set - they look quite separated from each other
generate_tsne_molecular_similarities(train_dataset, valid_dataset, test_dataset)
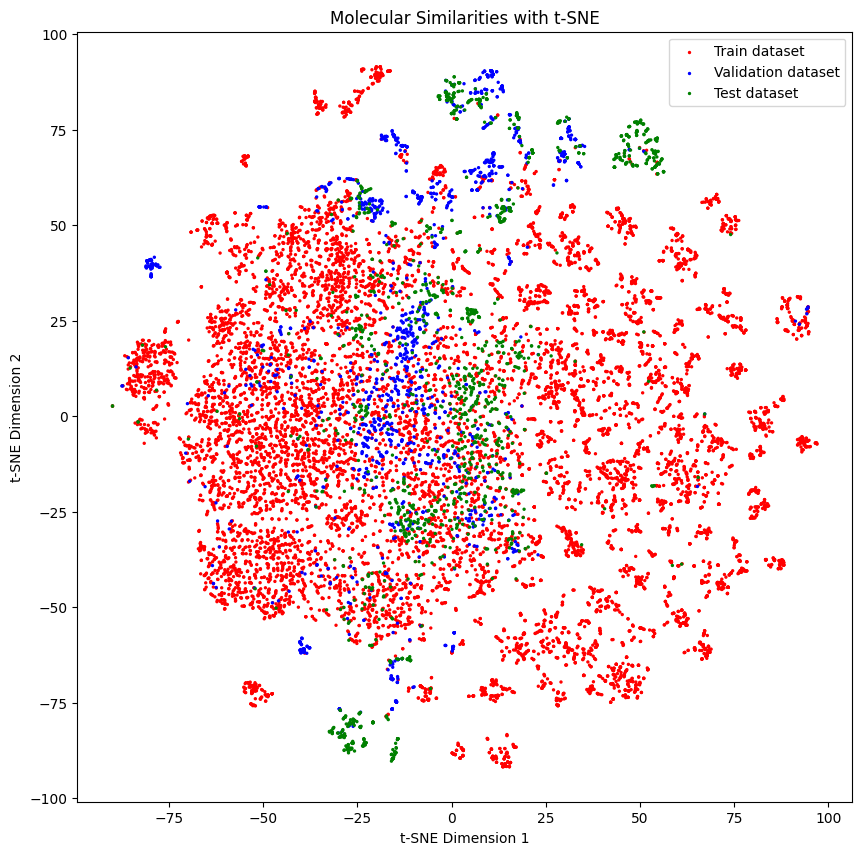
Then you can train them with a model. Here we use a random forest classifier. The performance is not as good as with the stratified splitter, but it is still quite good.
from deepmol.compound_featurization import MorganFingerprint
from deepmol.models import SklearnModel
from deepmol.metrics import Metric
from sklearn.ensemble import RandomForestClassifier
from sklearn.metrics import accuracy_score
MorganFingerprint().featurize(train_dataset, inplace=True)
MorganFingerprint().featurize(valid_dataset, inplace=True)
MorganFingerprint().featurize(test_dataset, inplace=True)
rf = RandomForestClassifier()
model = SklearnModel(model=rf)
model.fit(train_dataset)
model.evaluate(test_dataset, [Metric(accuracy_score, name="accuracy")])
({'accuracy': 0.7899159663865546}, {})
Let’s try with a lower threshold and see what happens
train_dataset, valid_dataset, test_dataset = similarity_splitter.train_valid_test_split(dataset, frac_train=0.8, frac_valid=0.1, frac_test=0.1, homogenous_threshold=0.10)
generate_tsne_molecular_similarities(train_dataset, valid_dataset, test_dataset)
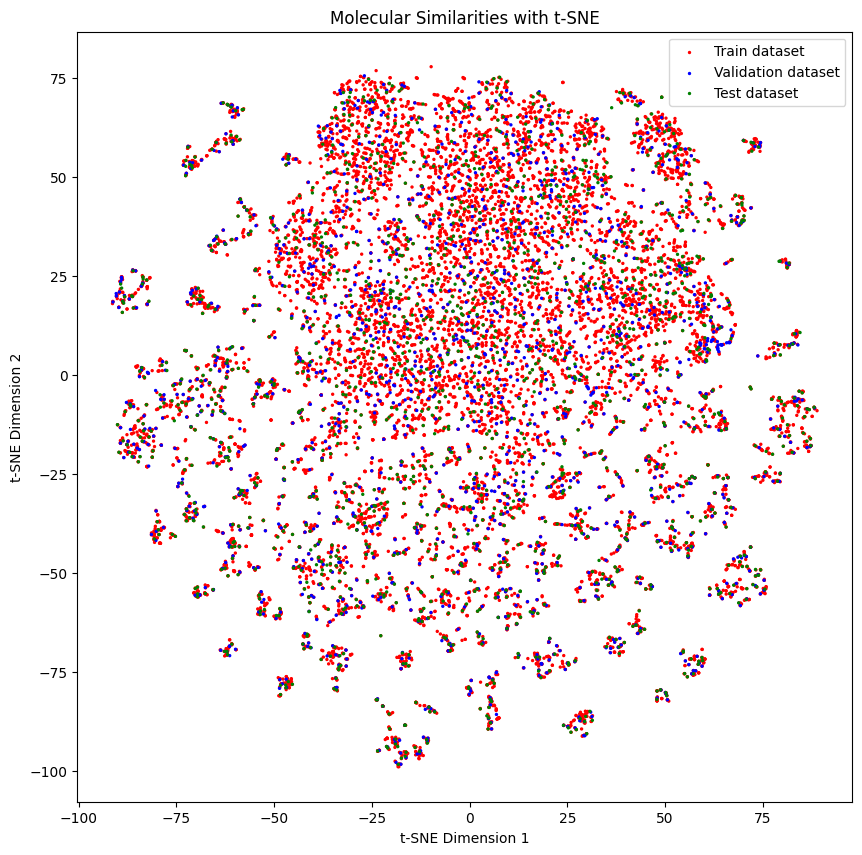
Molecules don’t look as separated as before, and of course, the performance is better than before.
from deepmol.compound_featurization import MorganFingerprint
from deepmol.models import SklearnModel
from deepmol.metrics import Metric
from sklearn.ensemble import RandomForestClassifier
from sklearn.metrics import accuracy_score
MorganFingerprint().featurize(train_dataset, inplace=True)
MorganFingerprint().featurize(valid_dataset, inplace=True)
MorganFingerprint().featurize(test_dataset, inplace=True)
rf = RandomForestClassifier()
model = SklearnModel(model=rf)
model.fit(train_dataset)
model.evaluate(test_dataset, [Metric(accuracy_score, name="accuracy")])
({'accuracy': 0.9873949579831933}, {})
Scaffold Splitter
The scaffold splitter splits the data into train, validation and test sets. The data is split in a way that the scaffolds of the molecules in each set are different or the same, depending on the homogenous_datasets parameter. This is useful when we want to make sure that the molecules in the validation and test sets are different or similar in terms of substructures from the molecules in the training set.
Let’s see how it works and create a split with different scaffolds in each set
from deepmol.splitters import ScaffoldSplitter
scaffold_splitter = ScaffoldSplitter()
train_dataset, valid_dataset, test_dataset = scaffold_splitter.train_valid_test_split(dataset, frac_train=0.8, frac_valid=0.1, frac_test=0.1, homogenous_datasets = False)
Below we can see the distribution of the molecules in each set
As you might see, the distribution of the molecules in each set is a bit different.
generate_tsne_molecular_similarities(train_dataset, valid_dataset, test_dataset)
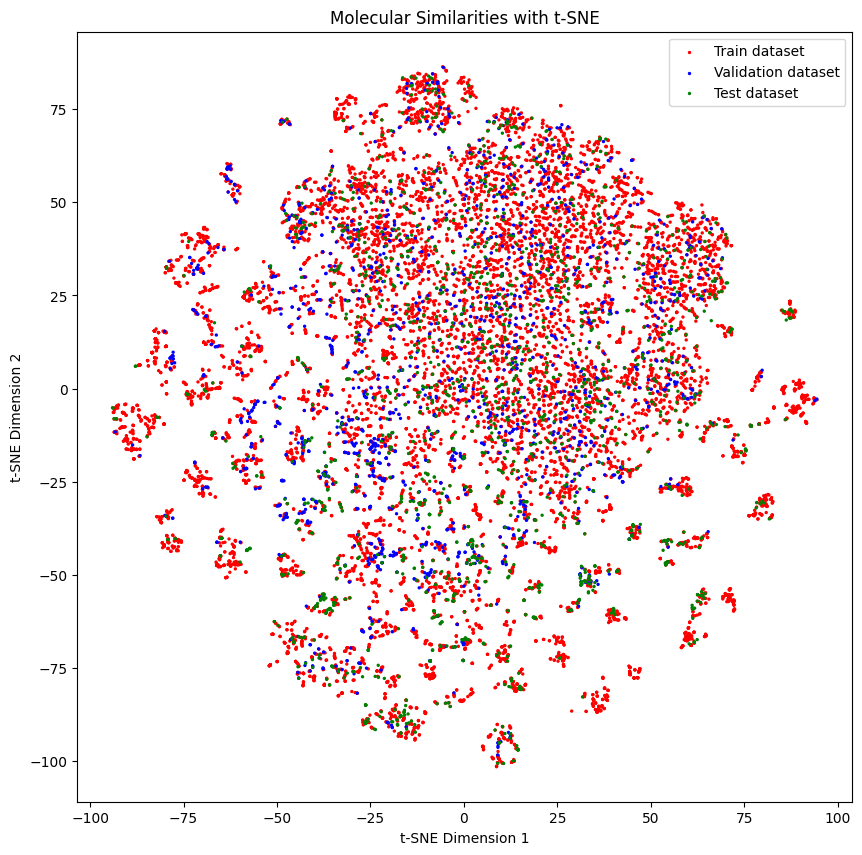
Let’s see how it works when training a model
from deepmol.compound_featurization import MorganFingerprint
from deepmol.models import SklearnModel
from deepmol.metrics import Metric
from sklearn.ensemble import RandomForestClassifier
from sklearn.metrics import accuracy_score
MorganFingerprint().featurize(train_dataset, inplace=True)
MorganFingerprint().featurize(valid_dataset, inplace=True)
MorganFingerprint().featurize(test_dataset, inplace=True)
rf = RandomForestClassifier()
model = SklearnModel(model=rf)
model.fit(train_dataset)
model.evaluate(test_dataset, [Metric(accuracy_score, name="accuracy")])
({'accuracy': 0.9663865546218487}, {})
Let’s make things a bit easier and create a split with similar scaffolds in each set
train_dataset, valid_dataset, test_dataset = scaffold_splitter.train_valid_test_split(dataset, frac_train=0.8, frac_valid=0.1, frac_test=0.1, homogenous_datasets = True)
generate_tsne_molecular_similarities(train_dataset, valid_dataset, test_dataset)

from deepmol.compound_featurization import MorganFingerprint
from deepmol.models import SklearnModel
from deepmol.metrics import Metric
from sklearn.ensemble import RandomForestClassifier
from sklearn.metrics import accuracy_score
MorganFingerprint().featurize(train_dataset, inplace=True)
MorganFingerprint().featurize(valid_dataset, inplace=True)
MorganFingerprint().featurize(test_dataset, inplace=True)
rf = RandomForestClassifier()
model = SklearnModel(model=rf)
model.fit(train_dataset)
model.evaluate(test_dataset, [Metric(accuracy_score, name="accuracy")])
({'accuracy': 0.9807923169267707}, {})
As you see, the performance is a bit better than before.
Butina splitter
The Butina splitter splits the data into train, validation and test sets. The data is split in a way that the molecules in each set are different or similar according to a clustering method called Butina, depending on the homogenous_datasets parameter. This is useful when we want to make sure that the molecules in the validation and test sets are different or similar from the molecules in the training set.
from deepmol.splitters import ButinaSplitter
butina_splitter = ButinaSplitter()
train_dataset, valid_dataset, test_dataset = butina_splitter.train_valid_test_split(dataset, frac_train=0.8, frac_valid=0.1, frac_test=0.1, homogenous_datasets = False)
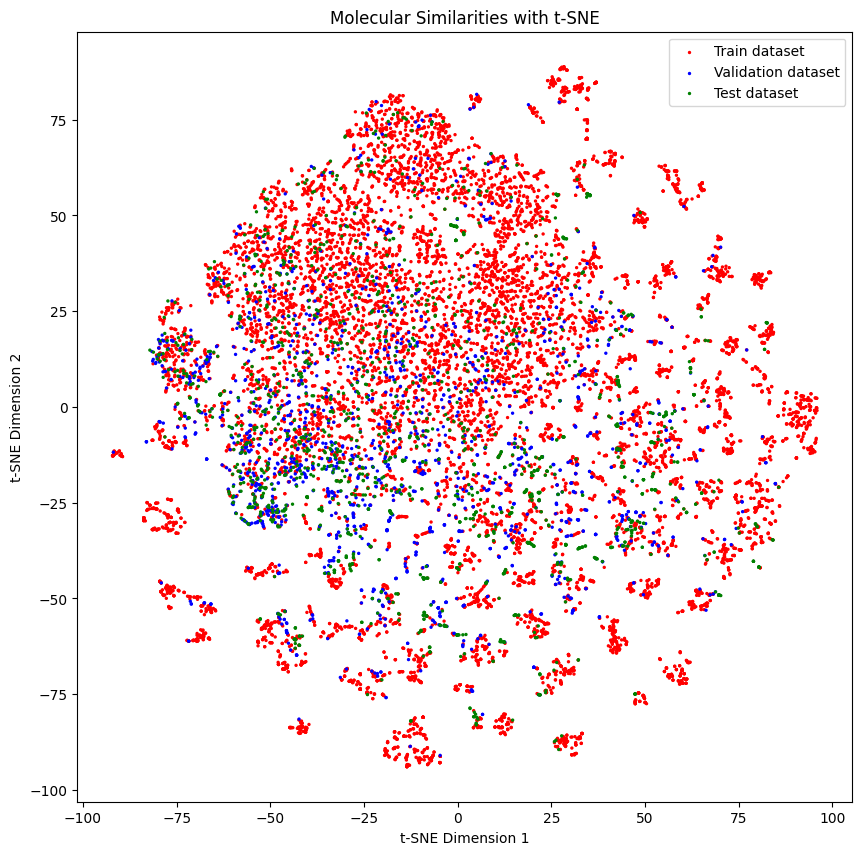
Below we can see the distribution of the molecules in each set
As you might see, the distribution of the molecules in each set is a bit different.
generate_tsne_molecular_similarities(train_dataset, valid_dataset, test_dataset)
from deepmol.compound_featurization import MorganFingerprint
from deepmol.models import SklearnModel
from deepmol.metrics import Metric
from sklearn.ensemble import RandomForestClassifier
from sklearn.metrics import accuracy_score
MorganFingerprint().featurize(train_dataset, inplace=True)
MorganFingerprint().featurize(valid_dataset, inplace=True)
MorganFingerprint().featurize(test_dataset, inplace=True)
rf = RandomForestClassifier()
model = SklearnModel(model=rf)
model.fit(train_dataset)
model.evaluate(test_dataset, [Metric(accuracy_score, name="accuracy")])
({'accuracy': 0.9147659063625451}, {})
Let’s make things a bit easier and create a split with similar molecules in each set
from deepmol.splitters import ButinaSplitter
butina_splitter = ButinaSplitter()
train_dataset, valid_dataset, test_dataset = butina_splitter.train_valid_test_split(dataset, frac_train=0.8, frac_valid=0.1, frac_test=0.1, homogenous_datasets = True)
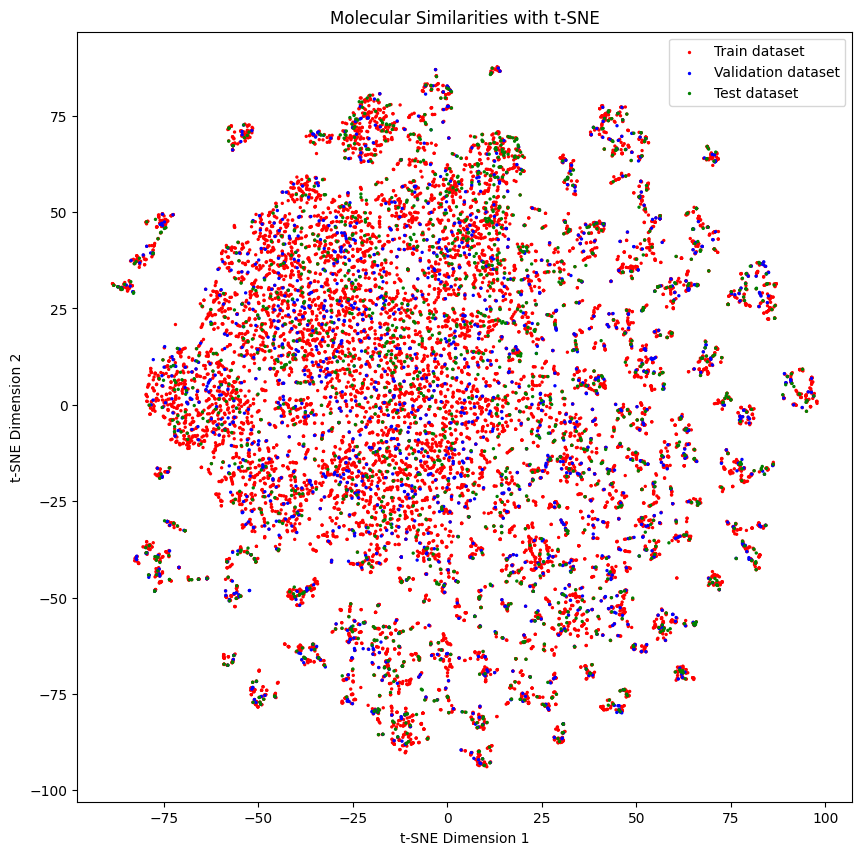
Below we can see the distribution of the molecules in each set
generate_tsne_molecular_similarities(train_dataset, valid_dataset, test_dataset)
from deepmol.compound_featurization import MorganFingerprint
from deepmol.models import SklearnModel
from deepmol.metrics import Metric
from sklearn.ensemble import RandomForestClassifier
from sklearn.metrics import accuracy_score
MorganFingerprint().featurize(train_dataset, inplace=True)
MorganFingerprint().featurize(valid_dataset, inplace=True)
MorganFingerprint().featurize(test_dataset, inplace=True)
rf = RandomForestClassifier()
model = SklearnModel(model=rf)
model.fit(train_dataset)
model.evaluate(test_dataset, [Metric(accuracy_score, name="accuracy")])
({'accuracy': 0.9617950272892662}, {})
As you see, the performance is a bit better than before.